



February 8, 2023
執筆者:Allison Strochlic、Merrick Kossack
米国食品医薬品局(FDA)が2022年12月9日に発行したドラフトガイダンス文書について、Emergo by UL、ヒューマンファクタリサーチ&デザイン(HFR&D)チームのリサーチディレクターであるMerrick KossackとAllison Strochlicが、2023年1月12日にウェビナーを開催しました。このドラフトガイダンス「Content of Human Factors Information in Medical Device Marketing Submissions」(医療機器市販申請のヒューマンファクタ情報)は、製造業者が510(k)、PMA(市販前承認)、その他の市販申請にどのようなヒューマンファクタ(HF)情報を含めなければならないかを説明しています。このドラフトが最終化されると、このガイダンスは2016年のドラフトガイダンス「List of Highest Priority Devices for Human Factors Review」(ヒューマンファクタレビューの最優先機器リスト)に優先します。ここで注意が必要なのは、今回の新ドラフトガイダンスは申請に最低限必要なHF情報を明記していますが、FDAが2016年2月に発行したHFEガイダンス「Applying Human Factors and Usability Engineering to Medical Devices」(ヒューマンファクタとユーザビビリティエンジニアリングの医療機器への適用)が、機器開発中に行う主なHFアクティビティの説明として依然として有効であるということです。
FDA HFEに関するウェビナー参加者からの主な質問
ライブ配信中にウェビナー参加者から、新ガイダンスの意味合いや、現在進行中の開発案件にどう適用されるのかといった多くの質問をいただきました。同じような疑問を持つ方も多いと思いますので、よくある質問とその回答を以下に記載します。
質問: HF報告書に、指定されたURRA(使用に関するリスク分析)ではなく、uFMEA(使用故障モード影響解析)を使い、その完全レポートを報告書に含める、または、別のリスクマネジメントドキュメントとして参照させることについてどう思いますか?
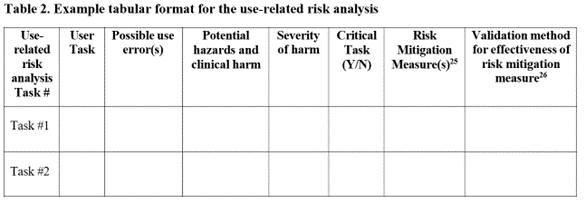
回答: 新ドラフトガイダンス(365~371行)に基づき、指定のとおりURRA情報を提出することを推奨します。uFMEAのすべてを提出する(または参照させる)よりも、できれば、ガイダンスのTable 2のフォーマットで最小限のコンテンツを提出するのがよいでしょう。URRAは(HFEに焦点を当てた)uFMEAの抜粋ですので、情報に関連性はありますが、FDAのHF審査官が重視するのは、uFMEAのデータの中でもクリティカルタスクの特定に関する部分の情報です。求められたとおりにURRAを提出すれば、審査官が主な使用に関するリスク情報にすぐにたどり着いて審査ができるため、不必要な遅れを生じさせることがありません。FDA新ドラフトガイダンス文書のTable 2は以下のとおりです。
質問: 「重大な危害」(serious harm)の新しい定義について詳しく教えてください。これに「死亡」が含まれるようになったことで、現在のクリティカルタスクのグレードやクラス分類が低くなることはありますか?
回答: Emergo by ULでは、新ドラフトガイダンスの「重大な危害」には、重傷“または”死亡が含まれると解釈しています。よって、死亡リスクがないからという理由でクリティカルタスクのグレードを下げられることはないとみています。この新ドラフトガイダンスでは「重大な危害」を「重傷と死亡の両方」を含むと定義していますが、これは「重傷または死亡のいずれか」を含むという意味であると、当社は考えます。重傷につながる可能性があるタスクは、死亡につながるリスクがなくても、クリティカルタスクです。
質問: 機器が欧州CEマーキングを取得していて、FDAのクリアランスや承認への申請をまだ提出していない場合、新ガイダンスやそれに含まれるフローチャートの文脈では、この機器は新しい機器または変更した機器とみなされることになりますか?
回答: そのような医療機器はFDAの観点からは新しい機器とみなされると当社では考えます。FDAは米国市場向け製品の認可や承認に責任を負っており、HFEやその他のガイダンスにおいても米国市場と米国住民が重視されているからです。ただし、米国外で実施したHF分析や評価の結果、そして市販後調査のデータなどがあれば、それをFDAへの申請に含めるべきです。クリティカルタスクがある場合、FDAから、関連する米国ベースのHF情報(例:米国人が参加したHFバリデーション(総括的)試験のデータ)も求められることが考えられます。
質問: 新ガイダンスと、FDAの別のガイダンス「Deciding When to Submit a 510(k) for a Change to an Existing Device?」(既存機器を変更した場合の510(k)申請をいつ提出するかの決定)はどのように関連しますか?
回答: 2017年のFDAガイダンス「Deciding When to Submit a 510(k) for a Change to an Existing Device?」は、既存機器に変更があったときに、新たな510(k)申請をいつ提出すべきかを製造業者向けに説明しています。2022年の新ガイダンス(282~283行)には、「[セクションV] は、FDAへの市販申請が必要な場合に、申請に適したHF情報を解説している」とあり、脚注27で「Deciding When to Submit a 510(k) for a Change to an Existing Device」を参照しています。これは、製造業者が2017年のガイダンスに基づいて、変更した機器で新たな510(k)を申請することを決定したら、2022年の新ドラフトガイダンスにあるフローチャートに従って、該当するHF申請カテゴリー(Submission Category)に関連する必須のHF情報を申請に含める必要がある、ということだと解釈できます。
新ドラフトガイダンスが(現在のドラフトでも今後最終化されても)、機器製造業者がどのHF情報をFDA申請に含めるかを決めるための助けになるでしょう。特に、既存機器の設計・デザインを変更した場合に役立ちます。大変重要な点は(本当に重要なのでこの文章を何度でも読み直してください)、機器のHF申請カテゴリーが1、2、3のどれであっても、機器が安全で効果的に使用でき、ユーザーのニーズに合ったものであるために、FDAは事業者に、2016年のプロセスガイダンスに沿った適切なHF適用を求めるであろうということです。
このウェビナー「New US FDA Guidance on HFE for Medical Devices」のライブ配信を見逃してしまった方やもう一度見たい方は、こちらで視聴いただけます。質問がある場合や、申請に含めるHF情報を決定する際の新ガイダンスの影響について評価が必要な場合は、ぜひご連絡ください。


Emergo by ULのエキスパートにご相談ください
お問い合わせありがとうございます。以下にご記入をお願いします。内容に応じて担当者が対応いたします。