



February 8, 2023
By Allison Strochlic and Merrick Kossack
On January 12, 2023, Merrick Kossack and Allison Strochlic, Research Directors of Emergo by UL’s Human Factors Research & Design (HFR&D) team, delivered a webinar to introduce and reflect upon a new draft guidance document issued by the US Food and Drug Administration (FDA) on December 9, 2022. The draft guidance document, Content of Human Factors Information in Medical Device Marketing Submissions, outlines what human factors (HF) information manufacturers should include with a marketing submission, including 510(k)s, PMAs, and others. Once finalized, this guidance will supersede the 2016 draft guidance, List of Highest Priority Devices for Human Factors Review. One key clarification is that while this new draft guidance specifies the minimum HF content in a submission, the HFE guidance published by FDA in February 2016, Applying Human Factors and Usability Engineering to Medical Devices, still stands in terms of outlining the key HF activities to perform during device development.
Key questions from our FDA HFE webinar attendees
Throughout our live webinar, we received dozens of questions from attendees regarding the nuances of the new guidance and how it might apply to their ongoing development efforts. Assuming others might have similar questions, we briefly share some of the most common questions and our responses below:
Question: In the HF report, what are your thoughts on including the complete uFMEA, or referencing a separate risk management document, rather than including the specified use-related risk analysis (URRA) content?
Response: Based on the new draft guidance (lines 365-371), we recommend submitting the URRA information as requested, ideally following the guidance’s Table 2 format for minimum content, rather than submitting (or, simply referencing) the full uFMEA. While the information is related – with the URRA being excerpted from the uFMEA (and HFE focused) – we believe the Agency’s HF reviewers are primarily interested in the subset of information from a comprehensive uFMEA that is related to identifying critical tasks. Submitting the URRA as requested will likely help avoid any unnecessary delays due to the reviewer being unable to quickly locate and focus on the key use-related risk information. See below for Table 2 as presented in FDA’s new draft guidance document.
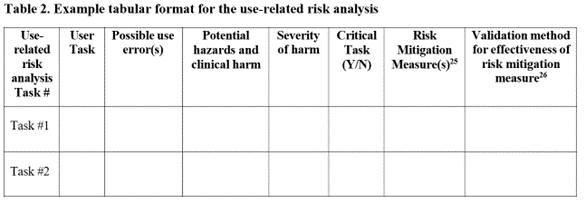
Question: Can you further explain the new definition of “serious harm?” Will this definition result in the downgrading or declassification of existing critical tasks since “serious harm” now includes death?
Response: Our interpretation of the new draft guidance is that "serious harm" includes serious injury OR death. Therefore, we do not expect critical tasks to be downgraded simply because they do not pose the risk of death. Note that although this new draft guidance defines “serious harm” as "includes both serious injury and death," we believe the intent is "serious injury OR death" noting a task can result in the former but not the latter.
Question: If a device has been CE-marked in Europe, but has not yet been submitted for market clearance or approval to the FDA, would the device be considered a new or modified device in the context of the new guidance and the included flowchart?
Response: We expect such a device would be considered a new device from the FDA's perspective, recognizing the FDA’s responsibility of clearing and approving products for the US market and their focus on the US market and US residents in HFE and other guidance. While you could, and should, include findings from HF analyses and evaluations conducted outside of the US in your submission as well as any post-market surveillance data, the FDA would likely also expect relevant US-based HF information (e.g., HF validation test data from US participants) if there are critical tasks.
Question: How does the new guidance relate to the FDA guidance Deciding When to Submit a 510(k) for a Change to an Existing Device?
Response: The 2017 FDA guidance titled Deciding When to Submit a 510(k) for a Change to an Existing Device is intended to help manufacturers determine when a change to an existing device warrants submission of a new 510(k). The 2022 new draft guidance (lines 282-283) states: "[Section V] describes the HF information that may be appropriate for submission to FDA in a marketing submission when one is required," and references footnote 27, which cites the Deciding When to Submit… guidance. We interpret this to mean that if you determine, per this 2017 guidance, that you need to submit a new 510(k) for your modified device, you should include the requested minimum HF information associated with the proper HF Submission Category per the flowchart in the 2022 new draft guidance.
We expect the new draft guidance – whether in its current form or when final – will help manufacturers determine what HF information to submit to the FDA, especially for those companies who are modifying an existing device’s design. Very importantly (so much so, you might want to re-read this sentence), regardless of whether your device ends up in HF Submission Category 1, 2, or 3, we believe that the FDA still expects manufacturers to perform proper HF work per their 2016 final process guidance to help ensure that a device can be used safely and effectively, and that it meets users’ needs.
If you didn’t catch the webinar live, or if you want to watch it again, you can do so: New US FDA Guidance on HFE for Medical Devices. Please reach out with questions, or if we can help you assess the impact of this new guidance when deciding what HF information to include in your submission.


Request more information from our specialists
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.