



August 29, 2024
By Ken Yoshida and Hinaho Ishikawa
As of April 1, 2024, the Japanese regulatory authorities, the Ministry of Health, Labor and Welfare (MHLW) and the Pharmaceuticals and Medical Products Agency (PMDA) require compliance with JIS T 62366-1:2022, Japan’s national Human Factors Engineering (HFE)/Usability Engineering (UE) standard for medical devices. If you are looking to market your medical device in Japan, you might be wondering what the Japanese regulatory HFE/UE expectations are. This article discusses the requirements in JIS T 62366-1:2022 and updates released by Japanese regulators.
Background on JIS T 62366-1
For an extended duration, the Japanese regulators did not mandate medical device manufacturers to apply HFE/UE to medical device development efforts. However, that changed with the release of JIS T 62366-1:2022, a Japanese Industrial Standard (JIS) that outlines the activities and the expectations for applying HFE/UE to medical devices. JIS T 62366-1:2022, released in 2022, is based on IEC 62366-1:2015+AMD1:2020 with technical requirements nearly identical to IEC 62366-1.
Following the release of JIS T 62366-1:2022, the Japanese regulators issued following documents:
- Handling of amendments to the Japanese Industrial Standards on usability engineering requirements of medical devices issued on September 30, 2022. This notification informs the industry that Japanese regulators’ mandate conformity with JIS T 62366-1:2022 starting April 1, 2024.
- Questions & Answers (Q&A) regarding compliance with JIS T 62366-1 issued on August 10, 2023. This Q&A is a supplemental document of the notification above, providing Japanese regulators’ responses to frequently asked questions about compliance with JIS T 62366-1:2022.
Two key takeaways are worth noting from these documents. First, the Japanese regulators emphasize the importance of updating standard operating procedures (SOPs) related to design and development processes, including risk management, to ensure compliance with JIS T 62366-1:2022. Therefore, manufacturers should conduct a comprehensive review of existing SOPs and update them based on JIS T 62366-1:2022. We encourage manufacturers to periodically update their SOPs to meet the evolving expectations of the Japanese regulators and other regulators across the globe.
Secondly, the Japanese regulators specify that, instead of JIS T 62366-1:2022, manufacturers are permitted to use internationally recognized standards (i.e., IEC 62366-1:2015) to demonstrate compliance with HFE/UE practices and expectations. This flexibility allows manufacturers to choose which standard they use when performing HFE/UE activities. If you opt to use IEC 62366-1:2015 for submissions to the Japanese regulators, it is important to understand the differences between IEC 62366-1:2015 and JIS T 62366-1:2022.
UOUP requirements for JIS T 62366-1
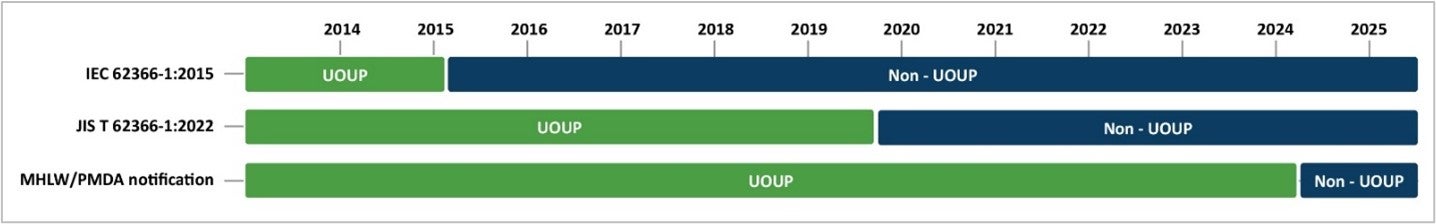
JIS T 62366-1 and IEC 62366-1 share the same content for the vast majority of topics. However, a notable difference between JIS T 62366-1 and IEC 62366-1 is the criteria for user interface of unknown provenance (UOUP). IEC 62366-1:2015 recognizes medical devices commercialized before February 24, 2015, as UOUP because the guidance was released on February 24, 2015. JIS T 62366-1:2022 considers medical devices released before October 1, 2019, as UOUP. Despite the UOUP criteria described in JIS T 62366-1:2022, the Japanese regulators’ notification indicates that medical devices released before April 1, 2024, can also be considered as UOUP. This means that not only devices released before October 1, 2019, but also devices released before April 1, 2024, appear to be allowed to follow the abbreviated UOUP process.
The required UOUP activities reflected in JIS T 62366-1:2022 are the same as reflected in IEC 62366-1:2015. The European MDR and Human Factors Engineering: Evaluation of User Interface of Unknown Provenance (UOUP) summarizes the five UOUP activities you need to perform.
Do you need to conduct an HF validation test in Japan?
A commonly asked question is whether the Japanese regulators accept HF validation test data collected outside of Japan as evidence that the device is safe to use as is by the intended users in the intended use environments and that the mitigations for highly-rated risks are effective. JIS T 62366-1:2022 has not explicitly mandated that HF validation test data must be collected in Japan. As such, Japanese regulators might be open to accepting HF validation test data collected outside of Japan. However, we recommend performing the following activities before leveraging HF validation test data collected outside of Japan for your Japanese submission:
- Understand users. Develop user profiles reflective of Japanese intended users to understand potential differences in how users in Japan interact with your device.
- Understand use environments. Develop use environment descriptions to identify potential differences between intended use environments in Japan and the simulated use environment in which you conducted the test.
- Analyze impact on use-related risk analysis (URRA). Assess how user and use environment differences may impact the use of your device. Revisit the URRA to identify whether the use of your device in Japan impacts the critical tasks or introduces new critical tasks.
- Evaluate labelling once translated. While there are controlled processes to verify the accuracy of labelling translations, we recommend assessing translated information and whether it impacts the validity of HF validation test data you already gathered.
Pending the identified differences and impact on highly-rated risks (i.e., critical tasks), conducting a supplemental, reduced-scope HF validation test in Japan might be deemed beneficial to ensure your medical device can be used safely and effectively by the intended Japanese users and in the intended Japanese use environments.
Conclusion
The Japanese regulators increased their focus on HFE/UE compliance, encouraging manufacturers to update their SOPs to align with JIS T 62366-1:2022. While the requirements and activities outlined in IEC 62366-1:2015 and JIS T 62366-1:2022 are comparable, they differ in UOUP criteria. As such, manufacturers should carefully assess how their devices are classified under IEC 62366-1:2015, JIS T 62366-1:2022 and the Japanese regulators’ notification.
Lastly, perform the aforementioned activities before determining/justifying forgoing an HF validation test in Japan. We strongly encourage manufacturers to fully understand the impact of local medical device use on critical tasks.
Please reach out if you need support bringing your medical device to the Japanese market. Emergo has a local team based in Japan, and we are dedicated in supporting medical device manufacturers in their efforts to secure regulatory approval in Japan.
Ken Yoshida is a Managing Human Factors Specialist and Hinaho Ishikawa is a Human Factors Associate at Emergo by UL.


X
Request more information from our specialists
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.
Please wait…
Characters remaining