



January 12, 2026
By Sarah Fitzgerald
Importance of Reviewing Warning Letters
By reviewing relevant U.S. Food and Drug Administration (FDA) warning letters, manufacturers can better understand areas of frequent concern related to quality management systems (QMS) and regulatory affairs. This can allow companies to focus on areas of highest risk as they continually assess and improve their internal procedures and processes.
Background on Warning Letters
The FDA issued 695 warning letters in 2025,[1] most related to drugs, tobacco products, or food products. Of those, about 8% (54) were issued for medical devices.
Overview of Medical Device Warning Letters
Most warning letters include multiple issues, however usually one issue appears to be central, based on the positioning within the warning letter (the first issue is generally considered to be the most significant) or occasionally the more detailed discussion within the warning letter. There were a similar number of warning letters issued primarily for regulatory issues as for QMS issues in 2025. The table below provides a summary of what Emergo experts believe are the main issues, based on review of these warning letters.
Table 1. Apparent Main CDRH Warning Letter Issues
Apparent Main Issue | 2023 | 2024[2] | 2025 |
|---|---|---|---|
Primarily Regulatory Reasons | |||
| Devices without marketing authorization | 3 | 10 | 7 |
| Marketing authorization insufficient / Does not cover current device or labeling | 8 | 9 | 7 |
| Investigational Device Violations (IDE, IRB authorization, monitoring, etc.) | 0 | 1 | 1 |
| Labeling Violations | 1 | 0 | 0 |
| FDA Establishment Registration and/or GUDID Registration Missing | 0 | 0 | 12 |
| Primarily Quality Management System (QMS) Reasons | |||
| Complaint, medical device reporting (MDR), and/or correction / recall insufficiency | 2 | 1 | 4 |
| Corrective and Preventive Actions (CAPA) insufficiency | 3 | 6 | 3 |
| Supplier / incoming product controls insufficiency | 1 | 4 | 2 |
| Design insufficiency (including Risk Management and Change Control) | 2 | 8 | 7 |
| Manufacturing process insufficiency (including non-conformance handling and final device acceptability) | 5 | ||
| Multiple QMS (unable to determine what appears to be the main issue) / Other | 4 | 2 | 0 |
| Other (Record maintenance, management oversight, internal audits, personnel qualifications and training, etc. | 0 | 0 | 3 |
| Testing Data Integrity & Reliability | |||
| Testing Issues (Testing facilities only: Data integrity and reliability) | 0 | 2 | 3 |
Details on Medical Device Warning Letters
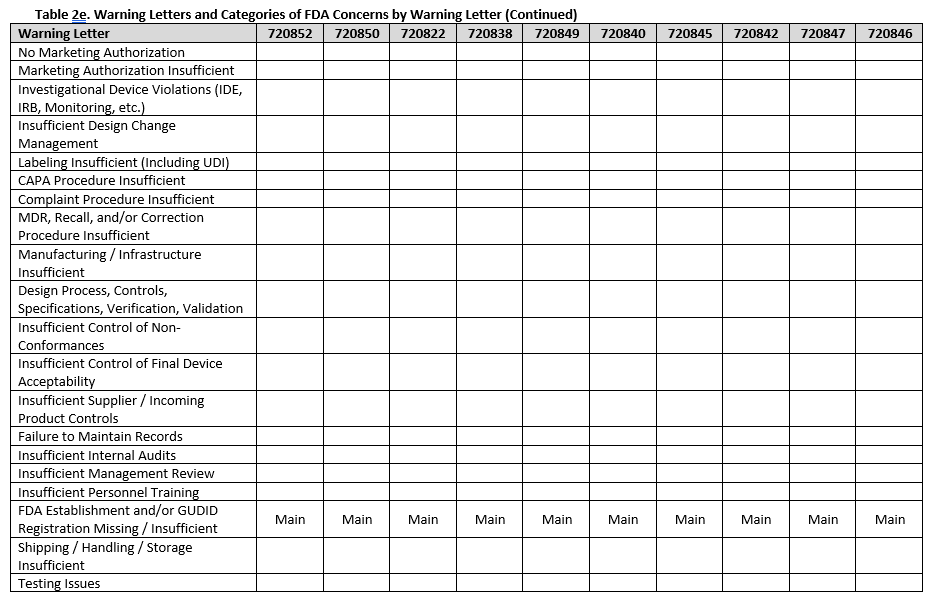
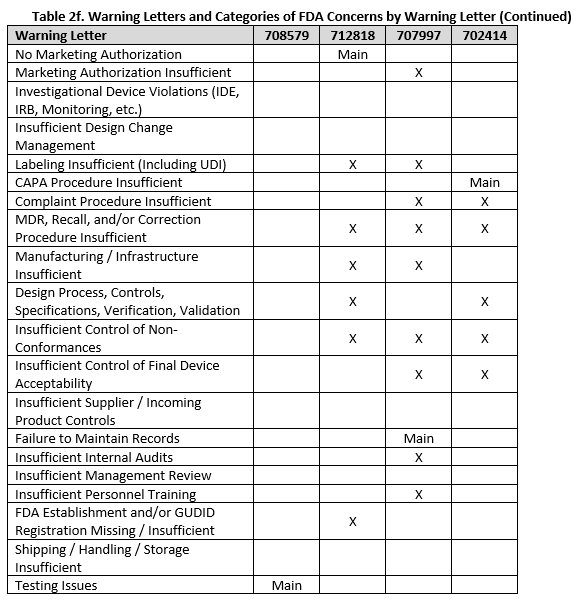
To fully understand FDA’s concerns, we need to look beyond the primary issue, as the majority of warning letters included multiple areas of concern. Tables 2a-f show all deficiency categories by warning letter. Note that for brevity, overall topics are grouped, so a warning letter may contain a different number of points than are provided, but the broad topics are addressed. Additionally, titles are minimized, therefore where something is noted as insufficient, this may range from no procedures covering a topic, to procedures not including certain required information, to the company not completely following a procedure.
[1] Posted warning letters as of January 8, 2026.
[2] The FDA CDRH annual report noted there were 44 warning letters issued, but a search of the FDA database on January 24. 2024 only located 43 which were analyzed.
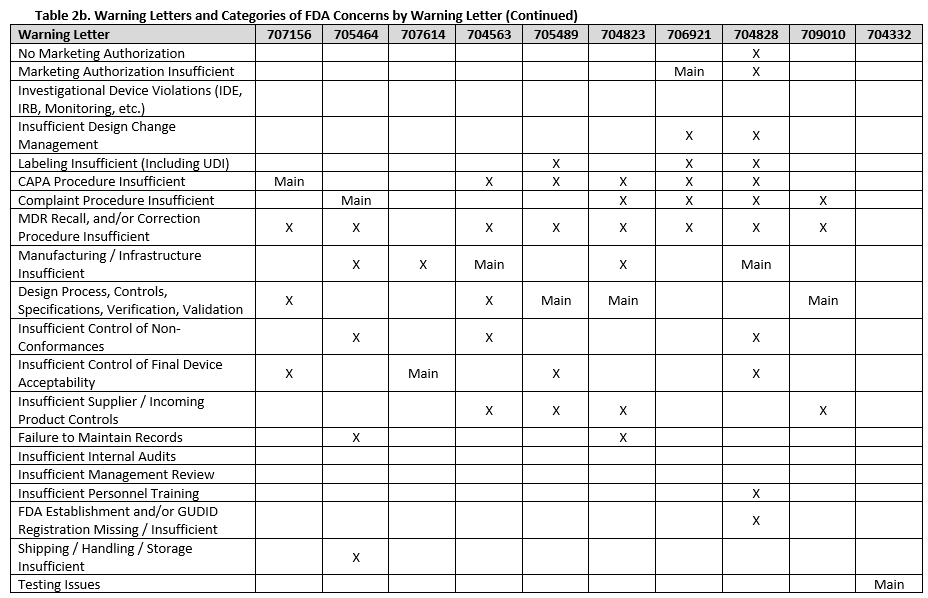
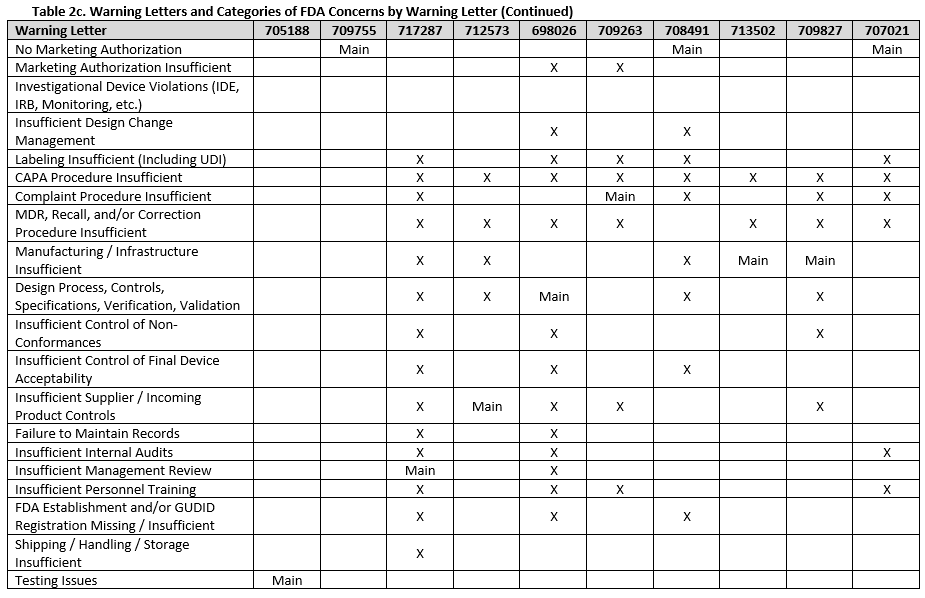
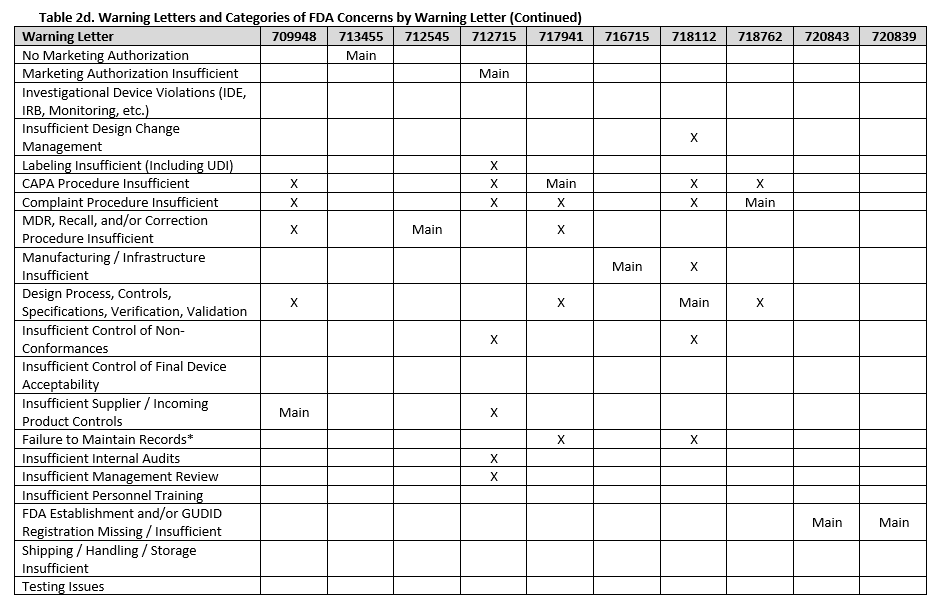
There were a few interesting considerations from our review of 2025 warning letters that are summarized below:
Twelve warning letters were issued for facilities manufacturing Class I, premarket submission exempt devices without appropriate FDA establishment registrations and device listings. All of these facilities manufacture Breast Binders (product code HEF), suggesting particular scrutiny on such products.
Three warning letters issued by CDRH were not for device manufacturers, but rather for testing laboratories working with animals for biocompatibility testing. Between the FDA news announcement and warning letters, the laboratories that the FDA has noted as producing unreliable data are: Mid-Link Technology Testing Co., Ltd, Sanitation & Environment Technology Institute of Soochow University Ltd., CCIC Huatongwei International Inspection Co., Ltd, Jiangsu Kerbio Medical Technology Group Co., and Palamur Biosciences Private Limited.
Risk management was a major topic of discussion for many of the violations this year, and was mentioned in many of the warning letters, including for the initial validation of device design.
Unique device identification (UDI) and the associated required registration in the Global Unique Device Identification Database (GUDID) were noted in several warning letters, emphasizing the continued importance of appropriate UDI labeling.
Categorization of the Warning Letters
Table 3 provides a summary of all deficiencies and calculates a percentage of the total of the medical device-related warning letters. This is done both for the overall and, because 12 of the warning letters were solely for registration deficiencies without appearing to have evaluated other potential deficiencies, with those 12 removed. Percentages are rounded to the nearest whole number.
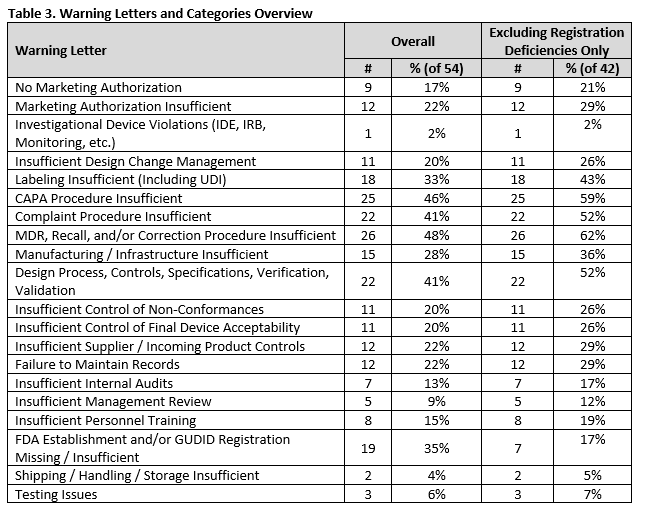
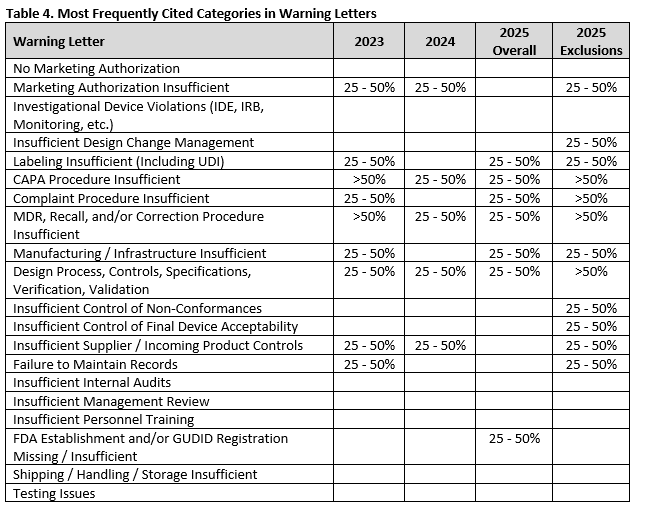
Table 4 summarizes the most frequently cited categories for 2023, 2024, and 2025, only noting those that are included in at least 25% of warning letters. Both the overall percentages and the percentages excluding those warning letters only including registration deficiencies (indicated as “2025 Exclusions”) are included in separate columns. The same categories are similar through the years, although it appears that the FDA may be commonly citing more types of issues in many warning letters as indicated by the greater number of categories cited in at least 25% of warning letters when letters with only registration deficiencies are excluded.
Concluding remarks
Manufacturers should pay attention to the procedures and processes within the categories that the FDA has found frequently deficient (as shown in Table 4) as they periodically assess these while working toward continual improvement.
In addition, a reminder that the QMS Regulation (QMSR, which incorporates by reference ISO 13845:2016) becomes effective on February 2.
Emergo experts are available to assist with QMS and regulatory considerations related to medical devices.




X
Request more information from our specialists
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.
Please wait…
Characters remaining