



December 11, 2023
Emergo by UL professionals shared an update on the EU Medical Devices Regulation (MDR, 2017/745) and In Vitro Diagnostic Devices Regulation (IVDR, 2017/746) on December 5 at the San Diego Regulatory Affairs Network (SDRAN) virtual evening program. It was an opportunity to relate our most recent regulatory insights on EU medical device and IVD compliance.
Development of MDR and IVDR
We shared a slide to remind the audience of the protracted time from the initial European Commission proposal in 2012 to the European Parliament and European Council amendments in 2014 and 2015, respectively, to the trilogue meeting’s consolidated compromise text in 2016 and the final publication of the regulations in 2017. We shared infographics and factsheets from the EC Communication Campaign to highlight various elements of the MDR, 2017/745 and IVDR, 2017/746. We also discussed legacy devices.
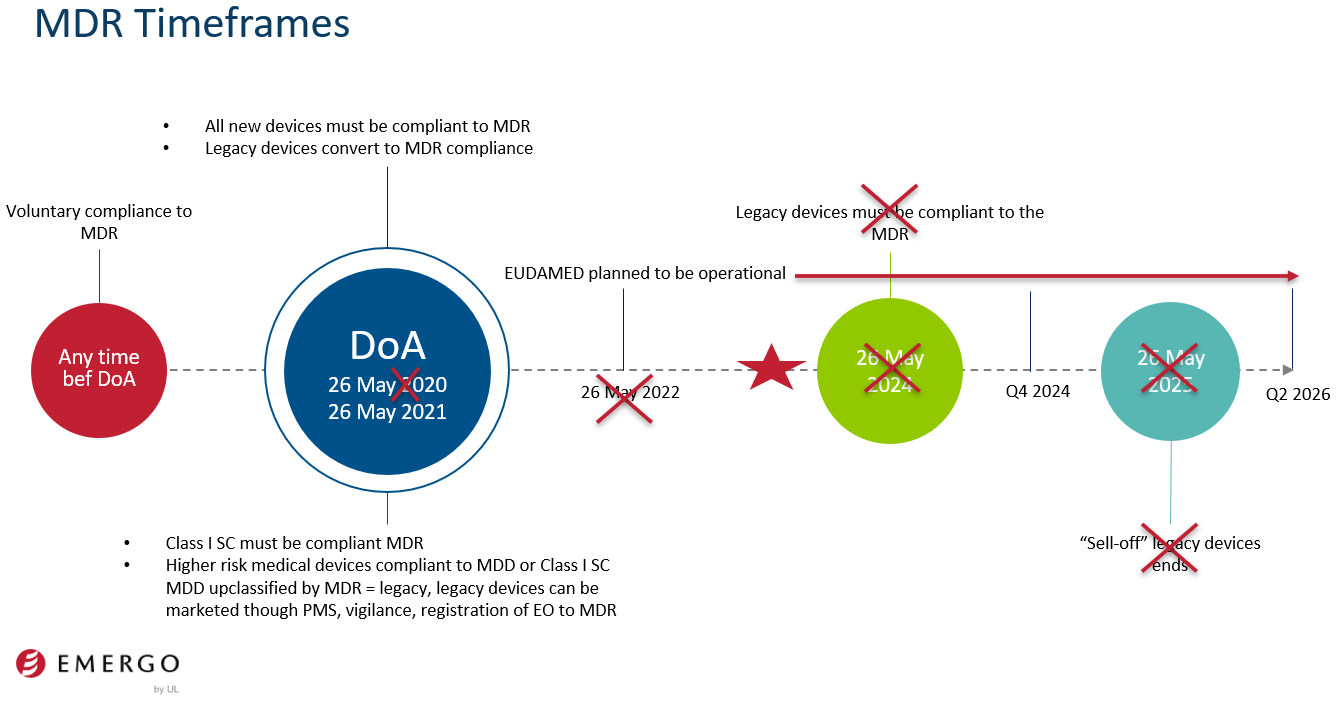
The frenetic developments from June to August 2022 that resulted in the draft regulation that amends the MDR and IVDR and culminated in Regulation 2023/607 were also shared. To illustrate the impact of these amendments (postponements), we shared a timeline slide:
Regulation 2023/607 sheds light on status of notified bodies and notified body applications
We covered Regulation 2023/607 and the conditions that need to be met to capitalize on the additional transition time. We also shared that there are currently 42 notified bodies designated under the MDR and 12 notified bodies authorized under the IVDR (as per the New Approach Notified and Designated Organizations (NANDO) database). We made recommendations related to the notified body strategy based on the following: capacity, designated to the regulations and appointed as a UK-approved body. In addition, we discussed the concept that now may be the time to change notified bodies.
We provided the latest data (through June 2023) on notified body applications, including the number of applications (13,177 and 1,155) and certificates (3,899 and 500) to the MDR and IVDR. We emphasized the significant time to obtain CE certificates (currently greater than 13 months) and the fact that the majority of applications were incomplete.
Strategies and goals to optimize reviews and common notified body observations
Some of our recommendations to optimize the review were presented:
- Assess the portfolio.
- Engage the notified body in discussion (especially through structured dialogues per MDCG 2022-14, item 15).
- Follow notified body guidance (including the individual notified body and Team-NB Best Practice Guidance (BPG) documents for the MDR and IVDR).
- Format accordingly (Annex II MDR and IVDR), standards in the Official Journal of European Union (OJEU).
- Review Medical Device Coordinating Group (MDCG) guidance.
- In addition, we shared that one of the sentiments expressed at RAPS Regulatory Convergence 2023 was that the manufacturer should try to tell the story about their devices: explain the background, provide the history and explain why the data is sufficient.
Our next 19 slides presented the common observations we have identified from examining many different notified body MDR and IVDR reviews.
While a lot was discussed, some highlights were:
- Device description needs to be comprehensive, and the Team-NB BPG should be used as companion guidance, including information about lifetime of the device.
- Many data elements on labeling need to be included, especially date of manufacture and medical device and near-patient testing.
- Standards and OJEU standards, and particularly packaging and transport.
- Benefit risk and the importance of compliance with EN ISO 14971.
- Clinical Evaluation Reports (CER) and Performance Evaluation Reports (PER) and identification of the relevant General Safety Performance Requirements.
- CER and clinical benefit, clinical development plan and intended use compared to indications for use.
- PER and IVD intended purpose (IVDR in Annex I 20.4.1 (c) and Annex II 1,1.1(c)).
- Post-market clinical follow-up (PMCF) general methods always required for medical devices (perhaps specific methods may not be).
- The import of the MDCG guidance for the Summary of Safety and Clinical Performance (SSCP) and Summary Safety Performance (SSP) for MDR and IVDR, respectively.
EUDAMED database update, legacy devices and MDCG guidance resources
We concluded with an update on the European database, EUDAMED; how to manage legacy devices; and all the tremendous resources available, including the extensive MDCG guidance documents. The last slide was an opportunity for us to opine on the importance of portfolio assessment, monitoring standards published in the OJEU, and MDCG and Team-NB guidance to facilitate the understanding of compliance requirements and deadlines. We also discussed that the CER and PER remain the number one notified body deficiency and that flexibility remains key.
We look forward to 2024. The deadline for medical device manufacturers to have an application with a notified body is May 26, 2024, per Regulation 2023/607, and to have a contract (agreement) with a notified body by September 26, 2024, per Regulation 2023/607. Manufacturers of legacy devices that are Class D IVDs must be compliant with the IVDR by May 26, 2025, to continue to access the EU market.


X
Request more information from our specialists
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.
Please wait…
Characters remaining